博文
【Science综述】电催化理论与实验相结合:深入探究材料设计
||
【摘要】
电催化在清洁能源转换中起着核心作用,为未来的技术实现了许多可持续的过程。本文综述了几种不同电化学转化过程中涉及水、氢和氧的电催化剂和相关材料的设计策略,用理论作为催化剂性能的合理化解释的手段。通过考察控制不同电化学反应的催化剂的共同原理,我们描述了一个阐明催化这些反应趋势的系统性框架,该综述也可作为新的催化剂开发指南,同时强调了需要解决的关键问题。通过扩展这个框架到新兴的清洁能源反应,例如过氧化氢生产、二氧化碳还原和氮气还原,其中开发先进的催化剂可以实现燃料和化学品的可持续生产。
【前言】
在保护环境的同时,为未来创造全球规模的可持续能源体系是当今人类面临的最重要的挑战之一。2013年全球能源需求量达18 TW,绝大多数(约80%)来自化石资源(煤炭、石油和天然气)。随着世界人口的不断增长和工业化的不断推进,预计全球能源需求将从2013年的18 TW进一步增加到2040年的24 TW(新政策)或26 TW(现行政策),相应的,二氧化碳排放量从2013年地32 Gt/每年增加到2040年的37或40 Gt。因此,能源供应尤其是与化石燃料使用相关的气候变化问题引起了人们的重大关注,通过转向太阳能、风能和水力发电等可再生能源来丰富能源来源方式,减少对化石燃料的依赖。
电力行业占全球能源的比例约为12%,因此可再生电力的更大突破对于全球能源结构的调整是很重要的。其他需要发展可持续发展途径的重点能源部门还包括交通和化学工业。2010年交通运输占全球能源的19%;运输能源的43%用于轻型车辆,其中电气化已经起到了减碳的作用,其余的57%用于商业运输的海运、航空、铁路以及电气化更具挑战性的重型道路车辆。预测显示在未来几十年,轻型运输能源需求会保持相对平稳;然而,2010年至2040年间,商业运输能源使用量预计将增长三分之二左右。由于化学燃料对于这个行业来说更为成熟和方便,所以人们对于开发这种燃料的可持续发展方式有着浓厚的兴趣。
在2010年,生产工业化学品的能源占全球能源的8%,几乎全部来自化石燃料。 为了满足全球对塑料和化肥等产品的需求,化工行业的能源消耗量预计在2010年至2040年间将增长约三分之二,例如氢(50 Mt/年)、过氧化氢(2.2 Mt/年)、乙烯(115 Mt/年)、丙烯(73 Mt/年)、甲醇(40 Mt/年)和氨(175 Mt/年),可持续的无化石燃料生产途径可以在减少二氧化碳排放方面发挥重要作用,同时提供日常生产全球所需的化学品。
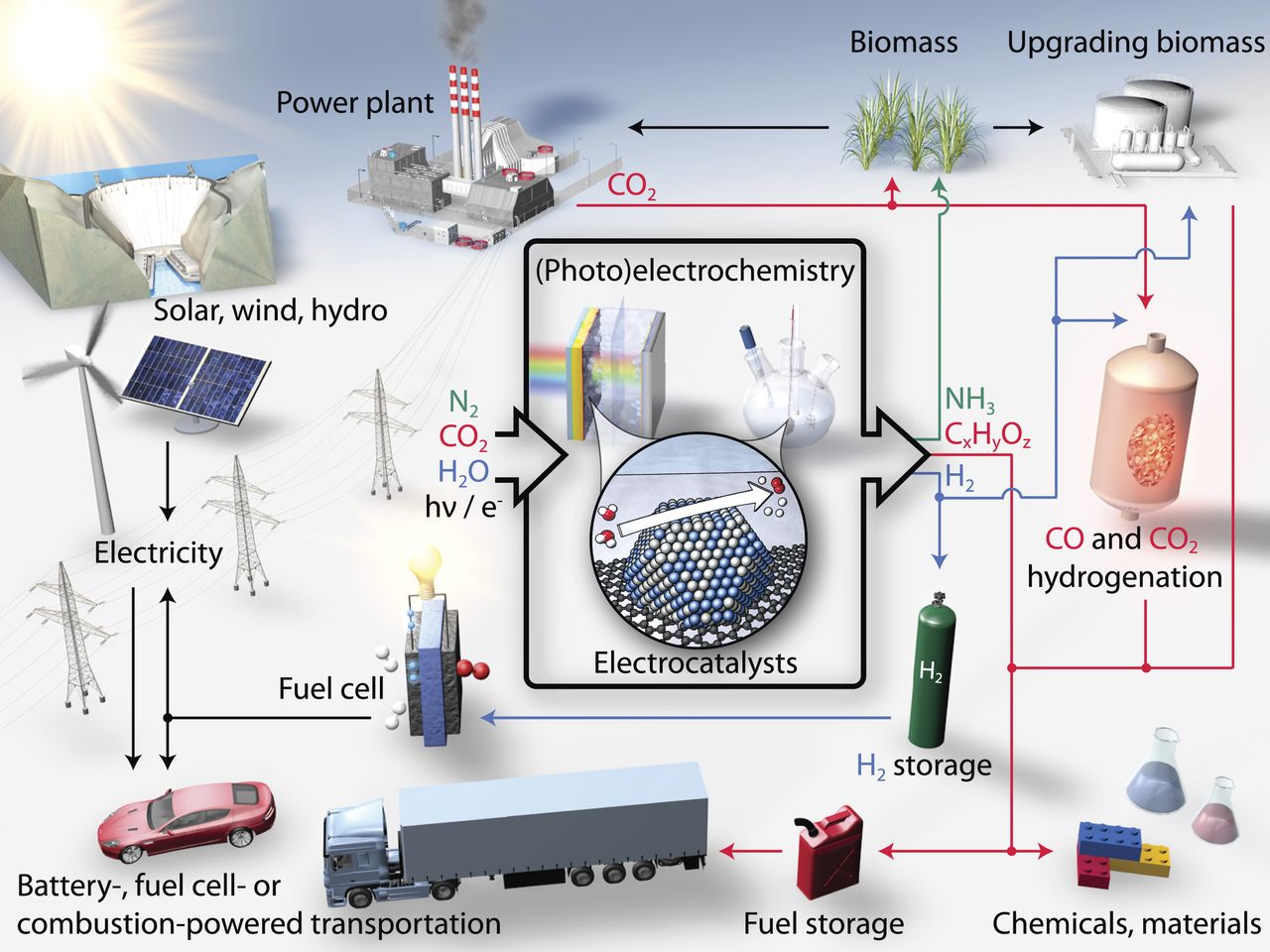
图1 基于电催化的可持续能源格局
图1显示了通过替代或与常规能源生产协同工作,生产重要燃料和化学品(包括氢气、碳氢化合物、含氧化合物和氨)可能的可持续途径。地球大气层提供了水、二氧化碳和氮气等通用原料,如果可以开发出具有所需性能的电催化剂,则可以通过与可再生能源耦合的电化学过程将这些原料转化为上述产物。例如,由析氢和析氧半反应组成的水分解反应可作为氢气的可持续来源已经引起了高度的重视。氢气是一种引人注目的能源载体,可用于燃料电池中产生清洁电力,发生氢氧化和氧还原反应将化学能转化为电能。过氧化氢是纸浆漂白和水处理行业的重要化学品,可用氧还原反应(ORR)来生产双氧水。从大气中或直接从源头捕获的二氧化碳,通过初步电还原将之转化为燃料、日用化学品、精细化学品、聚合物和塑料等的前驱体。同样,电还原可将氮气还原成氨,可以使肥料在使用地点和所需浓度下可持续地和局部地生产,从而消除了由于僵化的大规模集中的Haber-Bosch工艺所导致的分配成本。实现这一愿景的关键是开发改进的电催化剂,对所涉及的化学转化具有适当的效率和选择性。
通常有两种策略来提高电催化剂体系的活性(或反应速率):
(1)以“量”取胜:增加给定电极上活性位点的数量(提高分散度、增加活性位点暴露的数量)。
(2)以“质”取胜:增加每个活性位点的本征活性。(译者按,一语说中催化研究之要害,无出其右)

图2 催化剂开发策略的示意图
当然这两个策略并非互相排斥,可以同时用来提高催化剂的活性。同时,在不影响电荷传输和传质等其他重要过程的情况下,电极上能负载催化剂的量存在物理极限,因此(如图2)在高负载量时,实际中会观察到平台效应。另一方面,增加本征活性导致电极活性的直接增加,减轻由高负载量引起的输运问题;同等条件下,提高催化剂的本征活性,可以降低催化剂载量,这节约催化剂成本。催化剂活性可跨越许多数量级,性能优异的催化剂和不良催化剂之间的本征活性差异可以超过10个数量级,而高负载量催化剂和低负载量催化剂之间的差异可能只有1-3个数量级。(译者按,本征活性具有更广阔的提升空间)
近年来,电催化领域的论文出版数量迅速增加,表明电催化取得了很大进展。这篇综述重点讨论了几个电催化能量转换反应的典型案例研究,考察了最先进的催化剂并使用理论作为性能趋势合理化解释的手段。通过调研涉及水、氢和氧的多个反应,描述了电催化清洁能源转换趋势的框架。
首先介绍基于描述符方法的理论结果用于理解催化趋势,旨在建立一个选择少数且关键的催化剂表面性质的框架,但是仅这样一个描述符对于高活性可能是不够的。描述了这种相对快速、简单、直接的方法如何在近年来成功地实施以开发先进的催化剂。接下来的主要步骤是扩展建模能力,以不需要过多时间和资源的方式捕捉催化剂和电极-电解质界面的更复杂的特征性能。开发建模方法,使用最少的资源来快速准确地预测反应机理,评估大量催化剂和反应条件数据是未来的一个重要目标。在操作条件下,开发能够在原子和分子级描述电极-电解质界面的先进实验方法。在这一点上,可通过使用微动力学模型进行更详细的计算来提供当前理论的描述,以进一步提供这些类型的见解(例如反应速率和机理)。基于描述符的方法涵盖了大量体系,耦合单一体系的精细研究已经证明是有效的。未来努力推进理论和实验的融合将会更精细地描述表面上的催化作用。
【析氢/氢氧化反应】
需要活性催化剂来最小化驱动析氢反应(HER:2H+ + 2e– → H2)所需的过电位。HER是两个电子转移反应的经典实例,其中一个催化中间体H*(其中*表示电极表面上的位点),并且可以通过Volmer-Heyrovsky或Volmer-Tafel机理发生:
Volmer step: H+ + e– + * → H* (1)
Heyrovsky step: H* + H+ + e– → H2 + * (2)
Tafel step: 2H* → H2 + 2*
整个反应的速率很大程度上取决于氢吸附自由能△GH。如果氢气与表面键合太弱,吸附(Volmer)步骤将限制整个反应速率,而如果键合太强,解吸(Heyrovsky/Tafel)步骤将限制反应速率。因此,具有HER活性的催化剂的必要不充分条件是△GH ≈ 0。在适当覆盖度下,通过密度泛函理论(DFT)计算得到各种催化剂的△GH与实验测量的交换电流密度绘图,出现了“火山型关系”,定量说明了所谓Sabatier原理(图3A)。一个具有活性的催化剂会既不太强也不太弱的键合反应中间体。了解如何控制表面活性中间体的结合能是设计高性能催化剂的关键。
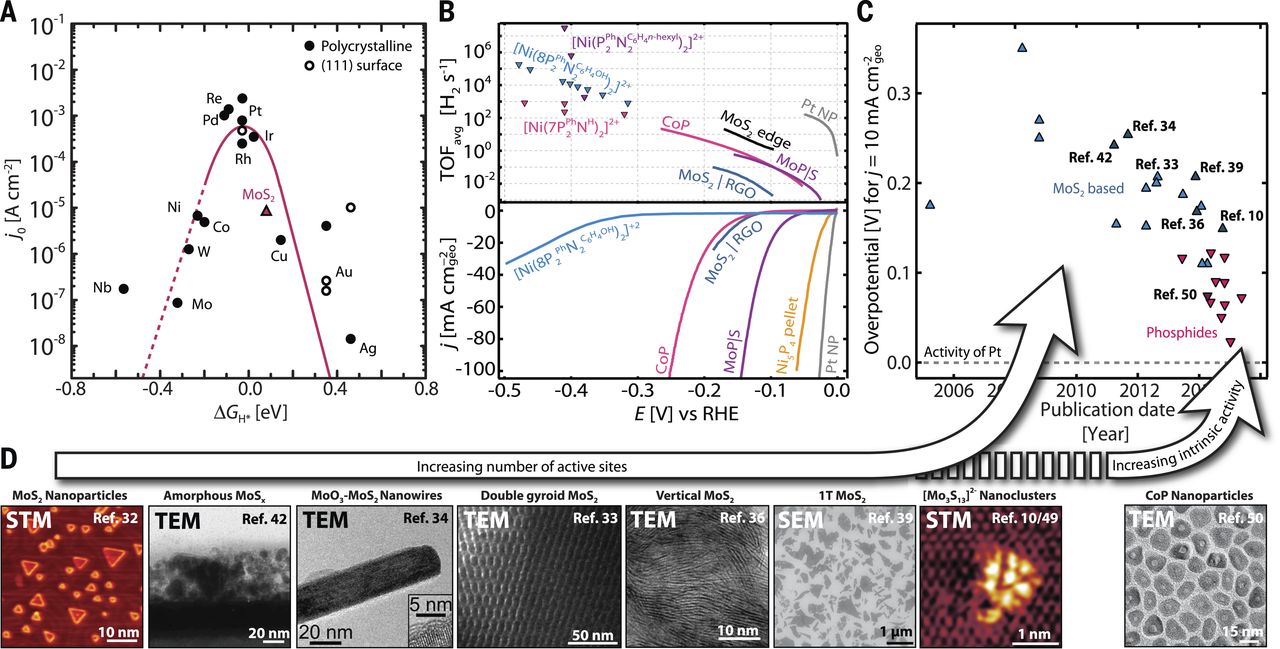
图3(A)金属和二硫化钼HER的火山曲线(B)TOFavg图,各种HER催化剂的线性扫描伏安图。(C)基于MoS2和磷化物HER催化剂的超电势的时间趋势。 (D)HER催化剂的代表性显微图像。
对于给定类别的催化剂材料,火山曲线可提供最佳催化剂的见解,但是火山曲线是简单地基于描述符模型,模型中还存在其他因素,这些因素是定量确定绝对反应速率所需的;例如,一类材料和另一类材料的动力学势垒大小是不一样的。这就是为什么尽管MoS2具有接近最佳值的△GH值,但是却观察到MoS2比图3A所示的贵金属具有更低的交换电流密度。对于给定的可逆氢电极(RHE),动力学势垒也可以作为pH的函数而改变,这导致观察到了电流密度对pH具有依赖性。尽管所涉及的过程(例如关于pH或动力学势垒)可能存在变化,但活性火山曲线并不左右移动,而是上下移动;这意味着描述符仍然可识别最佳HER催化剂的键合特征。然而,为了对这些效应进行全面的量化理解,计算电化学势垒的更详细和有效的方法需要涉及氢氧根离子和水合氢离子的质子转移反应,这是未来建模工作的一个重要目标。
铂非常接近氢火山型曲线的顶部,几乎具有热中性的△GH,是众所周知的HER性能最好的催化剂,在酸性溶液中可实现高反应速率而需要的过电位可忽略不计(图3,B 和C)。然而,铂的稀缺性和高成本可能会限制其广泛的应用。这引发了人们寻找可取代铂催化剂的地球上丰富含量的催化剂,而MoS2基的HER催化剂的发展就是理论指导发现和设计新型电催化剂的一个极好例子。
几十年来,MoS2被认为对于HER是惰性的。然而,受自然界氢化酶和固氮酶等的启发,在MoS2的Mo(10-10)边上进行了DFT计算,结果表明在50%的氢覆盖度下,其△GH为0.08eV,接近最佳值0 eV(图3A)。形成鲜明对比的是基面的△GH为1.92eV,这解释了体相MoS2不良活性的原因。这些计算启发大家暴露更多的MoS2边缘,如MoS2负载在碳黑上以暴露边缘位置,并随后在膜电极组装装置中检验。在〜175 mV的超电势下实现了几何面积归一化的电流密度为10 mA cm-2geo,这是当时报道的在酸体系中活性最高的非贵金属HER催化剂。
不久之后,实验证实MoS2的边缘确实是HER的催化活性位点。在Au(111)表面上沉积单层MoS2纳米颗粒时,使用扫描隧道显微术测量纳米颗粒面积和边缘长度,并且发现HER活性与MoS2周长成线性比例关系,而不是与MoS2表面积成比例(图3B和D)。 理论和实验研究的结合提供了关键的认识,即只有MoS2边缘是活性的,从而激发大家开发最大程度暴露MoS2催化剂边缘位点的方法。
纳米化MoS2催化剂是一个可实现最大程度暴露边缘的方法(图2)。为此,研究了具有双螺旋形态的三维介孔MoS2纳米结构(图3D)。双螺旋结构的纳米级弯曲最小化了基面的扩展,暴露了高密度的活性边缘位点。结果,双螺旋MoS2的所有表面位点的平均转换频率(TOFavg)超过了使用类似的硫化技术制备的MoO3-MoS2纳米线的2到4倍(图3D)。然而,双螺旋结构的缺点是从活性位点到导电基底的电子传输距离长,由于垂直于MoS2基面的电子迁移率低于面内电子迁移率,导致电阻损耗增加大约三个数量级。为了缓解这个问题,合成了垂直排列的MoS2纳米结构,它不仅暴露了大量的表面边缘位置,而且还使得电子传输到导电基底的能力更强(图3D)。
在催化剂开发中另一个有吸引力的方法是将纳米粒子分散在高表面积的载体上(图2)。 例如,在还原的氧化石墨烯(RGO)纳米片上制备MoS2纳米颗粒。 相对于不含RGO的合成,使用RGO做载体导致更好的分散和减少的MoS2纳米颗粒的聚集,边缘位点数量增加和电荷传输增强(图3B)使得催化活性优异。
通过锂离子嵌入到MoS2的范德华力间隙中来调节MoS2的电子性质也被作为增加HER活性的手段(图2)。锂嵌入化学剥离MoS2,从2H半导体多晶型物到1T金属多晶型物的相变,这是另一种手段调控催化剂活性(图2和3D)。有人提出1T-MoS2比同样制备的2H-MoS2的活性增强是由于活性边缘位点数量的增加以及电荷转移电阻的降低。另一项研究进一步提出,1T-MoS2的边缘不是主要的活性位置,而基面可能是催化活性的。最近,MoS2基面上的空位也表现出很高的活性,可以通过应变来调变MoS2片。
由于其高表面积(图3D),无定形钼硫化物也显示具有高HER活性。可以使用电沉积或湿化学合成而无需任何热处理来制备无定形钼硫化物,这使得它们对于需要避免高温硫化的某些应用是有潜力的。这样合成的无定形钼硫化物材料的组成接近于MoS3,但是在电化学中施加还原电位后,原位研究证明其表面转变为MoS2。非晶态硫化钼可以进一步掺杂过渡金属如Fe、Co和Ni,大大提高其活性。在酸性条件下,性能的提升主要是由于表面积的增加,而TOFavg在中性条件下得到提升。
多年来,广泛调控MoS2基催化剂的以增加活性位点的数目,效果是显着的,但是它们的整体电极活性仍然是有限的,因为通常只有一小部分位点(边缘位点)对反应速率有贡献(图3B到D)。这导致在表面上设计具有不协调的硫的分子簇,其类似于MoS2的边缘,例如MoS2四面体、MoIV-二硫化物和硫代钼酸盐[Mo3S13]2-复合物(图3D)。提升催化剂的负载量是增加活性位点数量的另一种方法,但是这种方法最终将导致质量和/或电荷输送的限制(图2)。这激发了具有较高本征活性的其他催化剂的发展,利用基于ΔGH ≈ 0的描述符的理论框架。其中包括过渡金属磷化物、硒化物、硼化物、碳化物和氮化物等,其中一些在10 mA cm-2geo时的超电势方面表现出与Pt更接近的活性。然而,在酸性条件下,由于非贵金属催化剂负载量大和表面积大,非贵金属体系在TOFavg方面仍然落后于Pt几个数量级(图3B到D)。在碱性介质中,还开发了优异性能的非贵金属催化剂,其中一些(例如Ni-Mo体系)在10 mA cm-2geo时的超电势较低。然而,这种体系的TOFavg值也大大低于Pt,与酸性非贵金属体系相似,因此有很大的提升空间。具有高TOFavg的均相催化剂也被开发出来了,尽管它们通常需要大的超电势才能达到可观的电流密度(图3B)。
在酸和碱体系中开发类似于铂催化性能的非贵金属HER催化剂仍然是重要的挑战。除了催化剂活性外,长期稳定性也是一个同等重要的指标,应该结合活性一起进行报道。评估催化剂耐久性的有用方法包括加速循环伏安测试、量化催化剂浸入电解质中的量以及使用薄膜催化剂形态。
氢氧化反应(HOR)和HER,除了反应是相反的,所涉及的反应步骤是相同的,在非贵金属催化剂的开发中受到的关注较少。根据理论,最佳的HOR催化剂也应该表现出ΔGH ≈ 0并位于图3A中同一火山的顶部。因此,预计Pt将成为酸中HER和HOR的最佳纯金属催化剂,实际上在实验中观察到这一点。然而,对于MoS2催化剂来说不是这样的,相对于HER,其显示出更差的HOR活性。理解贵金属和非贵金属催化剂HOR之间的差异仍然是研究前沿。一个区别涉及到催化作用中的覆盖效应:Pt的ΔGH对氢覆盖率的依赖性很小,而MoS2的ΔGH表现出显着的依赖性。表面氧化对于一些催化剂体系也可以起作用;例如用非贵重金属和金属合金,在HER条件下预期的金属表面与在HOR条件下可形成的金属氧化物/氢氧化物表面完全不同。在不同的操作条件下,准确地模拟预期表面结构和催化剂的化学计量在催化中是一个更广泛的挑战。因此,研究人员继续寻找具有金属导电性和几乎不变的ΔGH的地球含量丰富烦人物质,这为HER和HOR的高性能电催化剂的设计开辟了新的契机。
【氧还原/析氧反应】
四电子的ORR(O2 + 4H+ + 4e-→2H2O)需要先进的电催化剂来提高其速率和效率。一般来说,ORR涉及四个质子-电子转移,将氧气还原为水,对于燃料电池是理想的,或者是双质子-电子路径,可用于生产过氧化氢。四电子途径可以通过几种机制进行。直接四电子机制本质上可以是解离或缔合,这取决于氧在催化剂表面上的解离势垒。间接四电子机制首先涉及双电子途径到过氧化氢,然后进一步还原成水:
解离:O2 + 2*→2O*(4)
2O* + 2H+ + 2e-→2OH*(5)
2OH* + 2H+ + 2e-→2H2O + 2*(6)
缔合:O2 + *→O2*(7)
O2* + H+ + e-→OOH*(8)
OOH* + H+ + e-→O* + H2O(9)
O* + H+ + e-→OH*(10)
OH* + H+ + e-→H2O + *(11)
上述所有中间体的自由能都是在各种密堆积的金属表面上计算出来的,并且构建了理论ORR活性和ΔEO关系的火山型图,Pt靠近火山的顶部(图4A)。对于与氧结合过强的金属,其活性受到质子-电子转移到O*或OH*的限制。另一方面,对于结合氧过弱的金属,取决于所施加的电势,活性受限于质子-电子转移到O2*(缔合机制)或O2中O-O键的断裂(解离机理)。
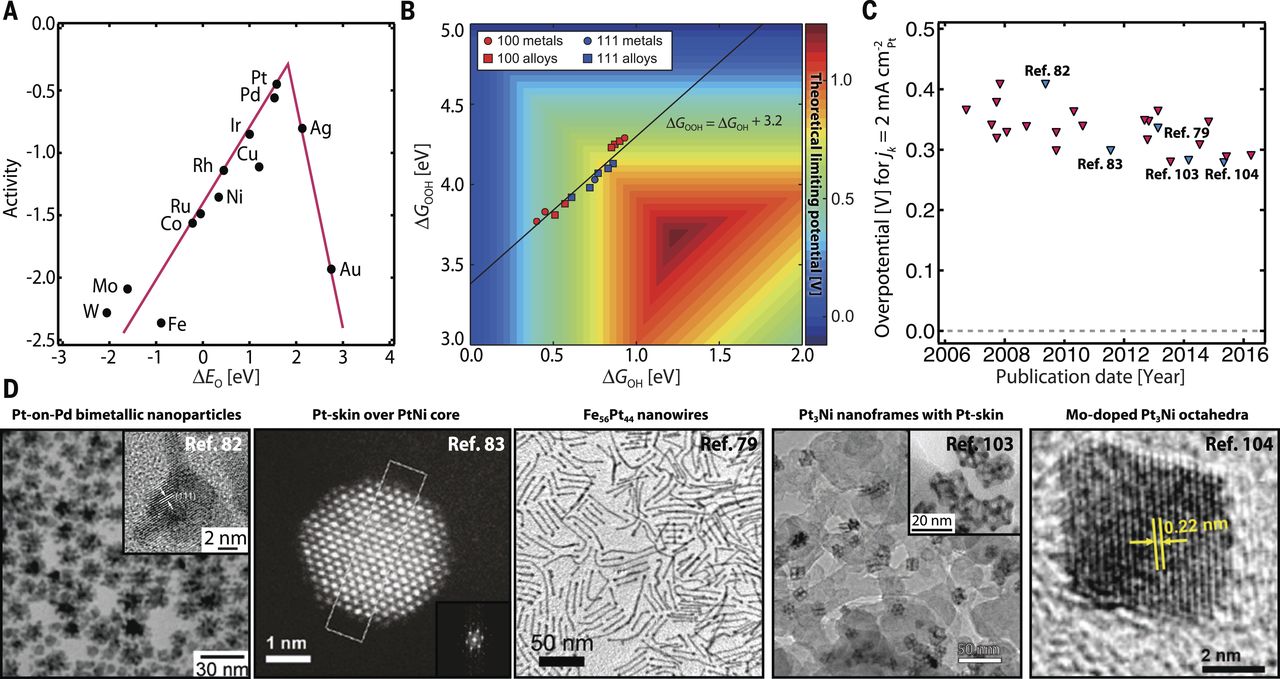
图4(A)金属的ORR火山曲线(B)金属和合金fcc(111)和(100)面的ORR理论极限电位图(C)Pt基ORR催化剂的过电位的时间趋势(D)ORR催化剂的代表性显微图像。
尽管图4A中呈现的火山型曲线类似于图3A中的HER/HOR所讨论的火山型曲线,但存在实质性差异。与具有一个反应中间体的HER/ HOR的情况不同,四电子ORR涉及多个中间体(OOH*、OH*和O*),其结合能是强关联的,并且由于标度关系(Scaling relations)不易解耦。实际上,标度关系(Scaling relations)ΔGOOH=ΔGOH + 3.2 ± 0.2 eV被发现普遍适用于面心立方(fcc)金属及其合金的(111)和(100)面(图4B)。由于OOH*与OH*之间的这种非理想的限制关系,即使是计算在具有最佳ΔEO的ORR火山型曲线顶部的催化剂也将具有0.3-0.4V的非零理论超电势。这也是观察到的超电位的起源,即使是最好的ORR催化剂,包括广泛研究的Pt基体系(图4C)。
在Pt基催化剂的开发中,相当大的精力已经用于形貌控制合成以调控ORR活性(图2)。在非吸附性电解质如高氯酸中,已知单晶Pt中低指数晶面的ORR活性遵循(110)>(111)>(100)的顺序。当使用吸附性电解质如硫酸等时,由于硫酸根阴离子强烈地吸附在(111)面,掩蔽了活性位点,所以(100)面比(111)面具有更高的活性。这些发现激发了具有不同形貌和暴露面的铂基催化剂的发展,包括纳米立方体、纳米管、纳米线、纳米枝晶和纳米笼(图4D)。
载体效应也可以在催化剂活性和稳定性方面发挥作用(图2)。目前,炭黑是铂基催化剂最常用的载体。然而,在高电位下炭黑的不稳定性与Pt-C相互作用弱是相关的,由此促使人们寻求更稳定的Pt催化剂载体,包括Ti0.7Mo0.3O2和掺锡的氧化铟,已经也显示出增强了催化活性。
合金化是提高Pt基催化剂的ORR性能的另一种广泛使用的策略(图2)。合金化可以降低Pt3M合金(M = Ni,Fe,Co,Ti)的顶层Pt的氧吸附能。Pt3Sc和Pt3Y在理论上被预测为有前途且稳定的Pt基合金ORR催化剂。这通过使用块体多晶Pt3Sc和Pt3Y催化剂的实验证实;相对于纯Pt,Pt3Sc催化剂的比活性提高了1.5至1.8倍,Pt3Y催化剂的比活性提高了6至10倍。这个策略已经扩展到包括碱土金属和镧系列。为了利用纳米结构的效应,还制备了尺寸选择的直径为4-9 nm的PtxY纳米颗粒,其中最好的是其质量活性比Pt纳米颗粒的质量活性高3倍。发现催化剂的活性随着平均Pt-Pt距离的减小而增加,表明合金核施加在表面Pt原子上的压缩应变提升了催化活性,进一步研究质量选择的PtxGd纳米颗粒。
另一种超过纯Pt的合金是Pt3Ni。制备了Pt3Ni(111)的延伸单晶表面,退火热处理和近表面区域的重构导致了最外层Pt的富集。这些所谓的Pt-skin结构表现出比Pt(111)高10倍的比活性和比商业化Pt/C催化剂高90倍的比活性。随后,从溶液中的PtNi3多面体的结构演变合成了三维Pt3Ni纳米框架(图4D)。这些具有大表面积且易于接近的纳米框架,相对于商业化Pt/C催化剂,即使经过10000次电位循环后,其比活性和质量活性分别提高了22和36倍。最近,报道用过渡金属掺杂Pt3Ni八面体进一步提高了它们的ORR活性(图4D)。特别是,与商业化Pt/C催化剂相比,掺杂Mo导致比活性和质量活性分别提高了81和73倍。计算结果表明,Mo在吸附氧的存在下优先占据表面顶点和边缘位置,形成较强的Mo-Pt和Mo-Ni键,稳定了Pt和Ni原子的溶解。尽管活性显着提高,但Mo掺杂的Pt3Ni系统仍然需要〜280 mV的过电位才能达到2 mA cm-2Pt,这远远低于ORR的平衡电位,为催化剂性能的提升留下了广阔的空间(图4C)。
如上所述,Pt是在酸体系中最好的HOR和HER纯金属催化剂,尤其是其微观可逆性:HOR和HER除了反应方向相反外,两种反应都涉及相同的步骤。根据相同的推理,人们可能认为Pt是最好的纯金属ORR催化剂,也是析氧反应(OER)的良催化剂;然而,这不是实验观察到的情况。一个原因是微观可逆性只适用于接近平衡的过程。当需要大的超电势来驱动两个方向的反应时,各个方向的催化要求可能会有很大差异。此外,在OER所需的高正电位下,包括Pt在内的金属通常会发生氧化,表现出与ORR条件下不同类型的表面。同样,催化研究的一个重要前沿领域是先进实验和理论方法的发展,能够快速准确地确定不同操作条件下催化剂的表面结构和化学计量。这样的信息对于充分了解反应动力学是必不可少的,并且使之与开发最好的催化剂放在同等重要的位置。
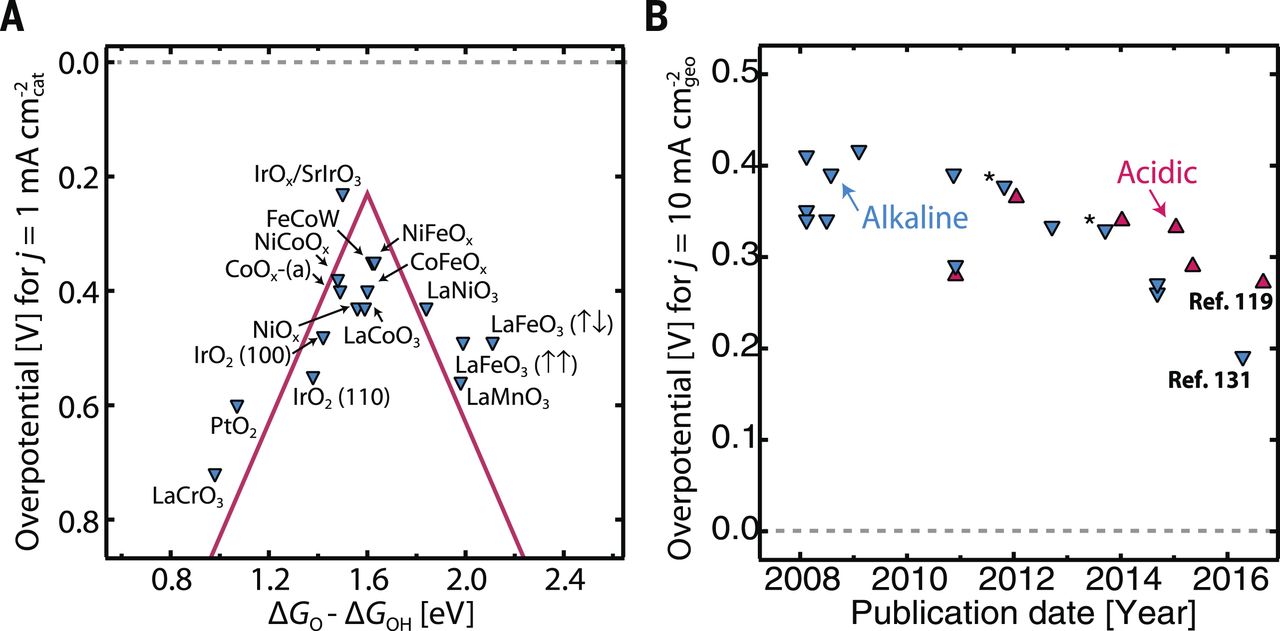
图5(A)金属氧化物的OER火山曲线(B)各种OER催化剂在酸碱中的超电势年代化趋势。
因为OER的催化剂通常是金属氧化物,所以使用ΔGO - ΔGOH作为描述符来构建各种金属氧化物表面(包括金红石、钙钛矿、尖晶石、岩盐和方铁锰矿氧化物)的OER的火山型曲线。当针对这个简单的描述符(图5A)绘制时,可以看出在1 mA cm-2cat处的实验超电势与理论上的超电势覆盖得很好。除此之外,更完整的模型将包含对所涉及的催化活性表面更精确的描述;例如,已经观察到图5A中的一些钙钛矿(化学式ABO3)在OER条件下经历A或B金属阳离子的浸出和表面非晶化。在没有完全阐明的所得催化活性表面的表面结构的情况下,图5A使用理想的结合能和化学计量终止的结构。在这些广泛的金属氧化物材料类型中研究的所有表面都被发现遵循OOH*和OH*之间的标度关系(Scaling relations)(ΔGOOH=ΔGOH + 3.2 ±0.2 eV),这与金属ORR的研究情况类似,再次阻碍了理论超电势为零的催化剂的发展。对于酸体系中的OER催化,IrO2是活性较好的金属氧化物催化剂,基于合适的反应中间体的结合能,理论上解释了其活性。基于在反应条件下的活性和稳定性,虽然这种催化剂已经在实验上证明是当今较好的OER催化剂之一,但是在活性方面远非理想的OER催化剂,并且在高氧化电位下不是完全稳定的。最近据报道,IrOx/SrIrO3薄膜显示出极高的OER活性(在10 mA cm-2oxide时为270 mV,归一化为氧化物表面积),并具有良好的稳定性。尽管薄膜的表面积低,但是相关的几何面积归一化活性也是超常的(图5B);利用优异的本征活性与提供更高表面积的方法(图2)可以使整体电极活性进一步得到提升。
由于贵金属如Pt、Ru和Ir的高成本和稀缺性,非贵金属氧化物催化剂如氧化镍、氧化钴、氧化锰和钙钛矿也被大力研究,虽然它们仅在碱性条件下才是稳定的。最近,制备了具有高表面积的三元FeCoW羟基氧化物电极(图5B),其在碱性电解质中对于OER表现出低过电位(在10 mA cm-2geo时为191 mV)。当归一化到催化剂表面积时,用于OER的许多非贵金属氧化物催化剂在碱性环境中拥有至少与贵金属体系一样的活性。这些催化剂中的一些在碱性电解质中对ORR和OER都有活性。通过研究一系列钙钛矿结构,ORR/OER活性与表面金属阳离子中的eg轨道填充之间建立了火山型关系,这可以作为活性的另一个描述符。在eg填充接近一致的情况下发现活性最佳,这时过渡金属-氧键具有很高的共价性。
最近,非金属催化剂在碱性条件下(例如杂原子掺杂的碳)也成为有希望的一类ORR/OER催化剂。通过掺杂更多的电负性原子如氮,在邻近的碳原子(C+)上产生净正电荷,这有利于氧的吸附和电荷转移,从而提高ORR/OER活性。这种策略也已经扩展到电负性比碳低的掺杂原子(例如硼),其产生类似的电荷位置(例如B+)以促进催化过程。为了利用不同掺杂原子的协同效应,碳催化剂的共掺杂也被证明是有效的。然而,非贵金属氧化物和非金属催化剂的典型缺点是它们在酸性环境下的稳定性差。总体而言,在酸性介质(例如质子交换膜燃料电池和电解质)中具有高活性和稳定性的地球含量丰富的ORR/OER催化剂的开发仍然是一个严峻的挑战。为了取得这方面的进展,需要对这些催化剂的工作状态及其活性部位的性质有更深入的了解,以适当的方式控制和调控它们的性质。 理论、计算研究和原位表征技术的结合将有助于解决这些关键问题。
尽管如图4A所示的热力学势垒火山型曲线有助于阐明ORR/OER催化的趋势,但仍需要向微动力学建模的火山型曲线方向发展。除了用于构建热力学火山曲线的基本步骤的反应能量之外,这种模型还需要每个基元步骤的动力学势垒大小。尽管对非电化学步骤的动力学势垒的计算已经被很好的理解,但是使用目前的技术来计算电化学动力学势垒(包括它们的电位依赖性)要困难得多。在很大程度上,这种困难是由于DFT固有的限制在恒定电荷的计算而不是恒定电位,这导致整个反应坐标的电位变化。一种规避这种限制的方案涉及外推至无限单胞的极限,其中单个电荷转移对模拟电势的影响可以忽略不计,因此在恒定电势下可获得反应能量和势垒。通过用逐渐增大的单胞进行计算,已经有可能推断出无限单胞尺寸的极限,其中在零驱动力下发现动力学势垒为0.26 eV,并且在Pt(111)上将OH*还原成H2O的电荷转移系数为0.5。
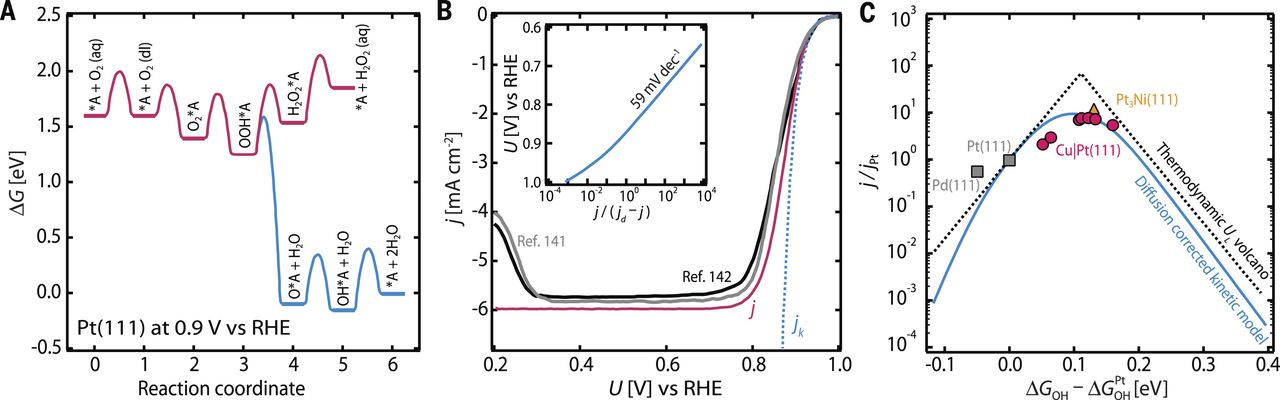
图6(A)在0.9V versus RHE下Pt(111)上的O2还原成H2O和H2O2的自由能图(B)在转速为1600 rpm时Pt(111)上的模拟极化曲线和动力学电流密度 (C)在0.9V versus RHE下模拟的动力学火山曲线。
进一步的研究已经将这些动力学参数纳入了ORR的所有四个耦合的质子-电子转移步骤中,成为一个稳态微动力学模型来研究Pt(111)。此外,模型中使用的反应能量和势垒是在明确的溶剂化双层水和在实验上和理论上发现的最稳定的覆盖率下计算的:具有2/3单层H2O和1/3单层OH的蜂窝(根号3×根号3)R30图案。如自由能图(图6A)所示,还考虑了双电子产物H2O2的形成。图6B比较了实验(黑色/灰色)和模拟(粉色/蓝色)极化曲线。考虑到扩散步骤(粉红色),对于起始电位和饱和电流密度,实验和理论之间达到了显着的一致性。最后,标度关系(Scaling relations)被用来在结合能描述符空间上的推广模型,从而创建一个由微动力学模型得到的理论火山曲线(图6C)。这项研究表明在复杂的电极-电解质界面多步电化学反应建模的进展,但未来需要更详细和有效的方法来确定电化学势垒,前面提到的一个重要前沿。
尽管多年来在酸和碱两者中开发ORR/OER催化剂方面进行了广泛的研究,但是大多数催化剂仍然需要0.25-0.4V的超电势以达到目标电流密度(图4C和5B)。上面提出的理论框架表明,这主要是由于ORR/OER中涉及的反应中间体之间的标度关系(Scaling relations)。克服这个特殊的限制需要去耦合不同中间体的结合能,例如通过稳定OOH*相对于OH*。尽管火山框架有助于阐明这一关键概念,但其实施将需要大量努力。对电极-电解质界面和相关动力学的更深入的理解将允许设计具有真正低过电势的ORR/OER催化剂。开发一种在已知标度关系(Scaling relations)周围设计催化剂的方法,只是创造近乎理想的ORR/OER催化剂体系的第一步,其将大大提高各种能量转换装置的效率。
除了涉及HER、HOR、ORR和OER的上述反应之外,还有许多其他新兴的能量转化反应相对较少探索。如果能够开发具有合适性能的电催化剂,其中几种可能会改变游戏规则。虽然这些反应可能涉及不同的反应中间体、机制和电子转移数量,但评估活性和选择性的描述符和火山型曲线的概念也是加速这些领域催化剂开发所需理解的第一个重要步骤。
【H2O2生产】
直接将氧气还原成过氧化氢(O2 + 2H+ + 2e- → H2O2)的电化学工艺的发展将是有利的,因为它可以直接耦合到可再生电力的方案中替代传统的能量密集型蒽醌工艺,以更安全的模块化分散的方式部署。由于过氧化氢在碱性条件下分解,所以通常在酸性环境中探索氧气电化学还原产生过氧化氢。
总的来说,由氧气产生过氧化氢涉及两个电子-质子转移耦合和一个反应中间体(OOH*),使得其与HER的复杂性类似:
O2 + * + H+ + e- → OOH*(12)
OOH* + H+ + e- → H2O2 + *(13)
因此,有可能找到具有最佳ΔGOOH的理论超电势为零的催化剂,其结合OOH既不太强也不弱。已经探索了诸如Pt、Ag、Au、Au-Pd合金、氮掺杂碳和分级多孔碳几种催化剂,发现在过氧化氢的生产中仅表现出适度的性能。与四电子路径相反,合适的电催化剂将需要对双电子具有高选择性。
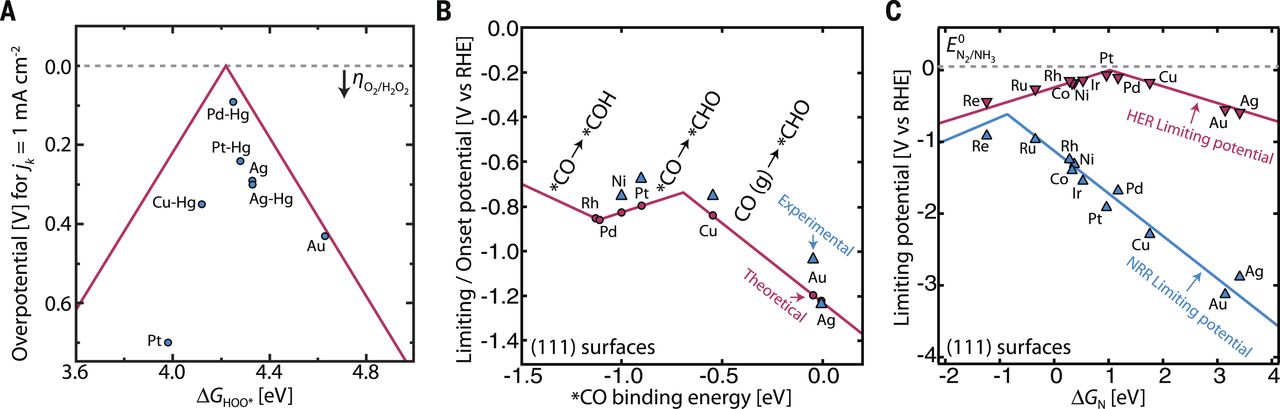
图7(A)在金属和合金上生产过氧化氢的火山曲线(B)金属二氧化碳还原的火山曲线(C)金属上的氮还原(NRR)与HER相比较火山曲线。
DFT计算已经建立了一个火山框架,该框架关联了理论超电势与氧气两电子还原过氧化氢的ΔGOOH,1 mA cm-2时的实验超电势叠加在这个曲线上(图7A )。对于强烈键合OOH*的金属,四电子ORR将比两电子路径占优势。另一方面,在OOH*弱结合的情况下,两电子和四电子火山型区域相互重叠,这表明在较弱的OOH*结合下,选择性产生过氧化氢的活性是折衷的。因此,在双电子火山曲线顶点发现了对过氧化氢具有高活性和选择性的最有希望的催化剂。理论预测的Pt-Hg、Pd-Hg和Ag-Hg合金在合成过氧化氢时不仅显示出非常高的质量活性,而且具有高选择性(> 95%)。在这项工作中的合理设计方法已经揭示了重要的概念,开始筛选和确定更有潜力的催化剂,特别是需要绕过汞的毒性。在基于热力学的框架的基础上,扩展它以更详细地了解动力学势垒和界面过程,在更广泛的材料和反应条件下,将有助于开发低超电位且对过氧化氢具有选择性的催化剂。
【CO2电还原】
另一个重要的能量转换反应涉及使用可再生能源将二氧化碳电还原为增值产品。像ORR一样,这是一个涉及许多不同的表面结合反应中间体的多电子还原反应。然而,与ORR(只有两种主要的最终产品,即水或过氧化氢)不同,有大量可能的二氧化碳还原产物,包括一氧化碳、甲酸盐、甲醛、甲烷、甲醇、C2+烃和含氧化合物;许多这些产品需要大量的质子和电子转移,并可能通过不同的中间体进行。因此,催化剂的选择性是一个主要挑战,由于大部分二氧化碳还原半反应的平衡电位接近于0 V versus RHE,使得HER成为竞争反应超过那些不想要的碳基副产品。因此,为了使二氧化碳还原在商业上可行,电催化剂将需要具有针对特定产品的高活性和高选择性。
在二十世纪八十年代,在各种各样的元素表面上研究了二氧化碳的还原。(1)一氧化碳(例如Au、Ag),(2)甲酸盐(例如Pb、Sn),(3)烃(例如Cu )和(4)氢(例如Pt、Ni)。在上述综合理论和实验框架的基础上,构建了一个初始火山曲线以了解二氧化碳还原的催化趋势。图7B由DFT计算组成,DFT计算的ΔECO与理论极限势相关,叠加的是形成甲烷/甲醇的实验开始电位,这是探测任一产物的起始电位。对于强结合CO*的金属来说,过电位是由CO*质子化到CHO*所决定的,而对于太弱结合CO*的金属,超电势由CO(g)对CHO*的质子化决定,其中CO解吸是竞争反应。对于甲烷/甲醇的形成,Cu被发现存在于靠近火山曲线顶部并具有最佳ΔECO的位置,由于标度关系(Scaling relations)的限制,理论上的超电势约为0.8V。考虑到每种产品所涉及的许多反应步骤和中间体,这并不令人惊讶,几种中间体可能以类似方式(例如在碳原子和金属表面之间)结合到金属上的C1物质。这指出了必须打破各种反应中间体之间的标度关系(Scaling relations),这对于生产甲烷/甲醇并增加几个数量级活性的二氧化碳还原催化剂来说是必要的,但也许是不充分的条件。理论指导表明,相对于CO*而言,增强CHO*(或者更确切地说,耦合的质子-电子转移到吸附的CO的过渡态能量)的结合能将使得CO*质子化到CHO *负电位较小,导致过电位显着降低。这可能通过许多策略来实现,包括合金化、解质添加剂、离子液体、束缚表面物种、促进剂和氢键供体或受体。
C2+物种的形成更复杂,因为C-C键形成是另一个需要考虑的反应步骤。两条途径已经确定。在一个途径中,一氧化碳首先被氢化,这使得C-C键更容易形成。 在另一个途径中,吸附的一氧化碳首先二聚化,这是形成乙醇和乙醛的重要中间体。考虑到二氧化碳还原反应的复杂性,需要更多的计算和实验工作来阐明潜在的反应机理和中间体。通过研究相关的一氧化碳还原反应也可以获得进一步的认识,这种反应类似于二氧化碳还原反应,但避免了形成甲酸盐可能引起的催化剂中毒效应。
【氮还原反应】
对于在室温和常压下进行电还原氮气(N2 + 6H+ + 6e- →2NH3)是受固氮菌中固氮酶的启发。早期的研究证明了这一过程在合成中的可行性,有助于促进包括Pt、Rh和Ru在内的催化剂的发展。与二氧化碳还原一样,氮还原反应涉及多个中间体,HER是一个主要的竞争反应,使其选择性是一个很大的挑战。迄今为止的实验结果表现出极差的性能(高超电势、低电流密度和低选择性),有很大的提升空间。
为了提供理论指导,使用DFT计算构建火山曲线,以将各种金属表面上的理论极限电势与ΔEN相关联(图7C)。这个框架再次提供了一种方法来理解金属催化剂用于该反应的一些关键点:金属键合氮过弱,会限制反应的第一步,N2吸附为N2H*;如果键合太强,会受限于金属NH*质子化形成NH2*(平面)或NH2*以NH3(阶梯表面)脱附。计算Ru、Rh、Mo和Fe等金属位于火山曲线顶部附近,氮既不太强也不太弱。不幸的是,由于中间体之间的非理想标度关系(Scaling relations),甚至在火山顶部附近发现的那些金属也表现出至少0.5V的理论超电势。对这一点建模表明,通过使N2H *比NH2*或NH*稳定来改善氮还原活性,这是必要条件,可能是不充分条件。
对于靠近火山曲线顶部的催化剂,HER也被发现是理论和实验上的竞争反应,因此不利于氮还原的法拉第效率(图7C)。据推测,Re、Sc、Y、Ti和Zr的平坦表面能够在相对于RHE的-1至-1.5V下进行氮还原,由于其对氮比氢的键合强,使得HER得到实质性抑制,但是结论是需要抑制HER的机制才能获得合理的表现。进一步的DFT研究还表明过渡金属氮化物,如VN和ZrN,在抑制HER的同时,在低起始电位对氮还原具有高活性。从最初的理论观点出发的材料设计策略是开发先进催化剂的第一个重要步骤。需要进一步的工作来阐明在电极-电解质界面处的原子尺度上的呃呃过程,包括溶剂、阳离子和阴离子的作用以及质子电子转移和N-N断裂的动力学。
【展望】
近年来,开发用于清洁能源转换先进电催化剂的兴趣日益浓厚。强调了初始方法的效用,该方法检验每个反应的基元步骤,以确定涉及的关键中间体、它们与表面的结合以及每个步骤的能量。对于每个反应,说明了这种方法如何帮助澄清已知催化剂体系中的关键限制以及这些知识如何导致开发先进电催化剂的成功策略。在大多数反应中,到目前为止限制催化剂成功开发的是吸附中间体的能量之间存在标度关系(Scaling relations)的限制。其中一个主要结论是需要一个新的催化剂设计范式来规避这些限制,特别着重于调整一个中间体相对于另一个中间体的稳定性。
一种策略是构建以不同方式结合不同反应中间体(和过渡态)的三维催化活性位点。例子可能包括合金化、掺杂或引入缺陷。或者通过选择性地稳定中间体(外部机制)可以规避标度关系(Scaling relations)。例如,尺寸的差异可以通过催化剂结构来区分,催化剂结构通过多个位点结合较大的中间体(如限域);如图2所示,通过将添加剂分子电解质或促进剂/配体到表面可以利用化学官能化(例如氢键)或物理性质(例如偶极强度)的差异。
还需要进一步的努力来阐明迄今为止仍不甚了解的电极-电解质界面的许多细节。当前的挑战包括溶剂、阳离子和界面附近的阴离子的原子分子级的描述,以及涉及质子/电子转移的关键基元步骤的动力学和反应势垒,在理论和实验中用更快速和更高效的方法来采集理解所需的信息是电催化研究的前沿,将会有更多的信息可以进一步指导开发接近理想效率和选择性的催化剂。
原文:Combining theory and experiment in electrocatalysis: Insights into materials design[J]. Science, 2017.
链接:http://science.sciencemag.org/content/355/6321/eaad4998
https://blog.sciencenet.cn/blog-3436572-1239259.html
上一篇:【电化学】浅谈塔菲尔动力学(Tafel Kinetics)
下一篇:牛津大学Compton教授—关于报道纳米材料电催化性能的思考